

ACTA CIENTÍFICA Y TECNOLÓGICA
REVISTA DE LA ASOCIACIÓN ESPAÑOLA DE CIENTÍFICOS Nº 2 AÑO 2000

APLICACIÓN DE DIFERENTES MODELOS DE REDES NEURONALES ARTIFICIALES (RNA), A LA SOLUCIÓN DE PROBLEMAS DE LA QUÍMICA ORGÁNICA, ESPECIALMENTE EN EL CAMPO DE LA QUÍMICA MÉDICA

1. Introducción
El trabajo que hoy voy a exponer aquí, se basa en una amplia aplicación de diferentes modelos de Redes Neuronales Artificiales (RNA), a la solución de problemas de la Química Orgánica, especialmente en el campo de la Química Médica. Esta nueva técnica de las RNA se ha integrado dentro de las ya clásicas del análisis de las relaciones cuantitativas entre estructura y actividad biológica (QSAR) o entre estructura y propiedades (QSPR), ambas técnicas, ampliamente utilizadas en Química Médica.
El trabajo que hoy voy a exponer aquí, se basa en una amplia aplicación de diferentes modelos de Redes Neuronales Artificiales (RNA), a la solución de problemas de la Química Orgánica, especialmente en el campo de la Química Médica. Esta nueva técnica de las RNA se ha integrado dentro de las ya clásicas del análisis de las relaciones cuantitativas entre estructura y actividad biológica (QSAR) o entre estructura y propiedades (QSPR), ambas técnicas, ampliamente utilizadas en Química Médica.
Mi trabajo consiste fundamentalmente en el aprovechamiento de la capacidad de predicción que tienen las RNA, predicción que se centra fundamentalmente en la actividad biológica de nuevos fármacos. Como sustrato o datos de partida, para la predicción de las propiedades de nuevas moléculas de la Química Orgánica, se parte de sus correspondientes fórmulas o estructuras gráficas, siempre desde un planteamiento puramente topológico.
2. Topología y Lenguaje gráfico de la Química Orgánica
Es sabido que la Química Orgánica ha desarrollado a lo largo de su existencia de casi dos centurias, un lenguaje propio para representar la naturaleza o constitución del objeto de su estudio, que es el de las moléculas que caen dentro de su campo de definición. Este campo o universo, es de dimensiones casi ilimitadas por lo que resulta de capital importancia disponer de un lenguaje suficientemente preciso para diferenciar no solo el inmenso caudal de moléculas conocidas (en la actualidad superior a 15.000.000), sino aquel otro caudal, constituido por moléculas aún desconocidas o de posible interés en cuanto a su preparación o síntesis. Así, en la actualidad y mediante la técnica de la química combinatoria y sus variantes, se pueden preparar o sintetizar a la vez - mediante un sólo proceso - cientos de miles de moléculas diferentes, no conocidas hasta la fecha.
Este lenguaje, que se concreta en una serie de fórmulas o estructuras gráficas, es suficientemente conocido, aún para aquellos, cuyo más reciente contacto con la Química Orgánica se remonta a su época de formación escolar. Sirva de ejemplo el clásico hexágono como fórmula o representación gráfica de la molécula de benceno.
Nicolás Turro, en una publicación emblemática, ha analizado el proceso intelectual mediante el cual se maneja este tipo de fórmula o representación gráfica de una molécula. Su hipótesis se basa en una interpretación más topológica que geométrica de la conectividad existente entre los átomos de una molécula. Para él, el químico piensa en términos topológicos y no en términos geométrico-euclídeos cuando interpreta o diseña una de estas estructuras gráficas, a pesar de que la apariencia de dicha representación gráfica tenga una naturaleza netamente geométrica. Según Turro, la mente se sirve de representaciones formales más abstractas para representar una molécula. Recurre para ello a la topología, que define un objeto matemático - tal que una molécula - como un espacio topológico. El espacio topológico correspondiente a una molécula - definido como un conjunto integrado por sendos subconjuntos de átomos y sus relaciones binarias - carece sin embargo de representabilidad gráfica. Esta sólo se consigue mediante la reinterpretación de dicho espacio topológico como un grafo, que sí puede representarse gráficamente.
Estas fórmulas o representaciones gráficas, tan profusamente utilizadas por los químicos orgánicos conservan, a pesar de su naturaleza geométrica, su esencia topológica. Esta se pone de manifiesto cuando en la fórmula de una misma molécula se alteran las dimensiones y/ó ángulos de los enlaces entre los átomos. Para el químico - que no para el lego - la nueva fórmula resultante sigue representando a la misma molécula, a pesar de que su geometría sea muy diferente de la inicial.
Mediante estas estructuras gráficas se pueden representar y manipular - sobre el papel o el ordenador - no sólo moléculas reales sino otras ficticias aún no preparadas en el laboratorio. El objetivo de mi trabajo consiste en predecir las propiedades de estas moléculas ficticias, con el fin de ahorrarle al químico el trabajo de sintetizar aquellas moléculas, cuyas propiedades no van a ser de interés para él.
3. Predicción
La predicción o la profecía, según Wittgenstein, son elementos clave en el desarrollo de la Ciencia. Así, en la actualidad, el enfoque predictivo ha dejado de ser una actividad esotérica para convertirse en un instrumento de gran potencialidad, en campos tan diversos como la economía, medicina, política o en la ya clásica meteorología.
Pieza clave de un proceso predictivo es lo que se entiende por reconocimiento. Para que este reconocimiento tenga lugar, hace falta disponer de un conjunto de conocimientos - en este caso sobre estructuras de moléculas de la Química Orgánica - a modo de base de datos o memoria. Este conocimiento se adquiere por aprendizaje, a lo largo de nuestra existencia. Su contenido - se asume - no es la suma de una serie de representaciones locales, esto es cada estructura tendría su representación perfectamente localizada, sino más bien una representación distribuida uniformemente por toda la base de datos o memoria. Esta distribución del conocimiento lleva consigo una suerte de solapamiento de los contenidos. Este solapamiento hace posible la existencia de un proceso de generalización que, no consiste más que en la posible extensión del conocimiento previamente adquirido a otras estructuras desconocidas hasta el momento. El reconocimiento de una estructura - previamente conocida - presupone un mapeo o mapping entre los rasgos de esta estructura y los almacenados en la memoria. En el caso de tratarse de una nueva estructura y por tanto desconocida para el sistema - entra en juego el proceso de generalización - mediante el cual se extrapola el conocimiento adquirido a esa nueva estructura, siempre que ésta tenga algún punto en común con las estructuras previamente almacenadas en la memoria. Rumelhart y McClelland definen precisamente el funcionamiento de las RNA como un procesamiento distribuido en paralelo (PDP). Esta es una de las facetas más importantes de las RNA y a ella se debe precisamente su capacidad predictiva.
4. Redes Neuronales Artificiales (RNA) y su integración en CODES
Un proceso similar, es el que aplicamos - mediante nuestro programa CODES - a las estructuras químicas. Efectivamente, no solo se reconocen las estructuras sino que mediante un proceso de generalización se extiende el conocimiento de estructuras conocidas a otras semejantes pero desconocidas. Todo este proceso que - partiendo exclusivamente de estructuras gráficas - nos lleva a la predicción de las propiedades de nuevas moléculas, está basado en un amplio uso de RNA.
Es sabido que la Química Orgánica ha desarrollado a lo largo de su existencia de casi dos centurias, un lenguaje propio para representar la naturaleza o constitución del objeto de su estudio, que es el de las moléculas que caen dentro de su campo de definición. Este campo o universo, es de dimensiones casi ilimitadas por lo que resulta de capital importancia disponer de un lenguaje suficientemente preciso para diferenciar no solo el inmenso caudal de moléculas conocidas (en la actualidad superior a 15.000.000), sino aquel otro caudal, constituido por moléculas aún desconocidas o de posible interés en cuanto a su preparación o síntesis. Así, en la actualidad y mediante la técnica de la química combinatoria y sus variantes, se pueden preparar o sintetizar a la vez - mediante un sólo proceso - cientos de miles de moléculas diferentes, no conocidas hasta la fecha.
Este lenguaje, que se concreta en una serie de fórmulas o estructuras gráficas, es suficientemente conocido, aún para aquellos, cuyo más reciente contacto con la Química Orgánica se remonta a su época de formación escolar. Sirva de ejemplo el clásico hexágono como fórmula o representación gráfica de la molécula de benceno.
Nicolás Turro, en una publicación emblemática, ha analizado el proceso intelectual mediante el cual se maneja este tipo de fórmula o representación gráfica de una molécula. Su hipótesis se basa en una interpretación más topológica que geométrica de la conectividad existente entre los átomos de una molécula. Para él, el químico piensa en términos topológicos y no en términos geométrico-euclídeos cuando interpreta o diseña una de estas estructuras gráficas, a pesar de que la apariencia de dicha representación gráfica tenga una naturaleza netamente geométrica. Según Turro, la mente se sirve de representaciones formales más abstractas para representar una molécula. Recurre para ello a la topología, que define un objeto matemático - tal que una molécula - como un espacio topológico. El espacio topológico correspondiente a una molécula - definido como un conjunto integrado por sendos subconjuntos de átomos y sus relaciones binarias - carece sin embargo de representabilidad gráfica. Esta sólo se consigue mediante la reinterpretación de dicho espacio topológico como un grafo, que sí puede representarse gráficamente.
Estas fórmulas o representaciones gráficas, tan profusamente utilizadas por los químicos orgánicos conservan, a pesar de su naturaleza geométrica, su esencia topológica. Esta se pone de manifiesto cuando en la fórmula de una misma molécula se alteran las dimensiones y/ó ángulos de los enlaces entre los átomos. Para el químico - que no para el lego - la nueva fórmula resultante sigue representando a la misma molécula, a pesar de que su geometría sea muy diferente de la inicial.
Mediante estas estructuras gráficas se pueden representar y manipular - sobre el papel o el ordenador - no sólo moléculas reales sino otras ficticias aún no preparadas en el laboratorio. El objetivo de mi trabajo consiste en predecir las propiedades de estas moléculas ficticias, con el fin de ahorrarle al químico el trabajo de sintetizar aquellas moléculas, cuyas propiedades no van a ser de interés para él.
3. Predicción
La predicción o la profecía, según Wittgenstein, son elementos clave en el desarrollo de la Ciencia. Así, en la actualidad, el enfoque predictivo ha dejado de ser una actividad esotérica para convertirse en un instrumento de gran potencialidad, en campos tan diversos como la economía, medicina, política o en la ya clásica meteorología.
Pieza clave de un proceso predictivo es lo que se entiende por reconocimiento. Para que este reconocimiento tenga lugar, hace falta disponer de un conjunto de conocimientos - en este caso sobre estructuras de moléculas de la Química Orgánica - a modo de base de datos o memoria. Este conocimiento se adquiere por aprendizaje, a lo largo de nuestra existencia. Su contenido - se asume - no es la suma de una serie de representaciones locales, esto es cada estructura tendría su representación perfectamente localizada, sino más bien una representación distribuida uniformemente por toda la base de datos o memoria. Esta distribución del conocimiento lleva consigo una suerte de solapamiento de los contenidos. Este solapamiento hace posible la existencia de un proceso de generalización que, no consiste más que en la posible extensión del conocimiento previamente adquirido a otras estructuras desconocidas hasta el momento. El reconocimiento de una estructura - previamente conocida - presupone un mapeo o mapping entre los rasgos de esta estructura y los almacenados en la memoria. En el caso de tratarse de una nueva estructura y por tanto desconocida para el sistema - entra en juego el proceso de generalización - mediante el cual se extrapola el conocimiento adquirido a esa nueva estructura, siempre que ésta tenga algún punto en común con las estructuras previamente almacenadas en la memoria. Rumelhart y McClelland definen precisamente el funcionamiento de las RNA como un procesamiento distribuido en paralelo (PDP). Esta es una de las facetas más importantes de las RNA y a ella se debe precisamente su capacidad predictiva.
4. Redes Neuronales Artificiales (RNA) y su integración en CODES
Un proceso similar, es el que aplicamos - mediante nuestro programa CODES - a las estructuras químicas. Efectivamente, no solo se reconocen las estructuras sino que mediante un proceso de generalización se extiende el conocimiento de estructuras conocidas a otras semejantes pero desconocidas. Todo este proceso que - partiendo exclusivamente de estructuras gráficas - nos lleva a la predicción de las propiedades de nuevas moléculas, está basado en un amplio uso de RNA.
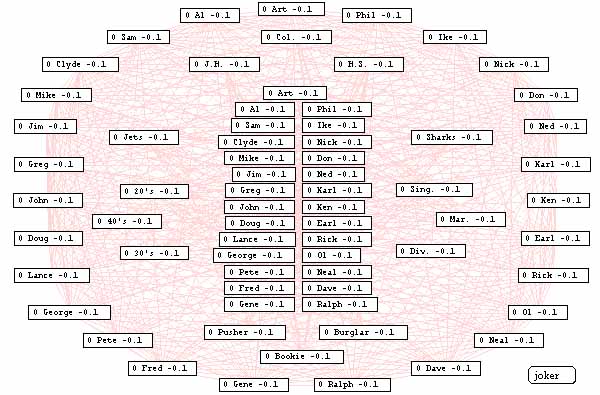
Fig.4.1. Estado inicial de la red, previo al experimento siguiente y que equivale a su estado de reposo. El fondo esta constituido por las conexiones excitatorias e inhibitorias que por su falta de actividad (-0.1) no están resaltadas. Este caso corresponde al clásico ejemplo de las bandas de los Jets y los Sharks, descrito por McClelland y Rumelhart. Cada integrante de la banda, se define por su instancia (unidades centrales) y por una serie de atributos personales (nombre, pertenencia a una de las bandas, edad, profesión, estado civil y estudios). Este modelo de red neuronal denomidado por sus autores como modelo de activación y competición interactivas (iac) sirve de fundamento al espacio neuronal creado por nuestro programa CODES.
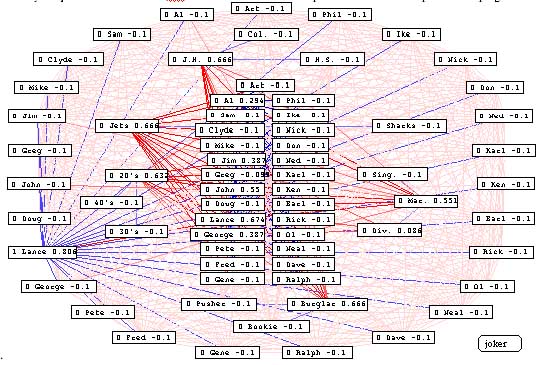
Fig. 4.2. Se ha activado el nombre Lance mediante una entrada externa de valor 1. En el transcurso del proceso, la actividad de Lance crece hasta alcanzar un valor de 0.806. Este valor activa por su parte - a través de las neuronas excitatorias coloreadas en rojo - los atributos de Lance. Los valores de actividad de estos atributos: Jets, JH, 20's, Burglar y Mar desactivan a su vez - por vía de las neuronas inhibitorias coloreadas en azul - el resto de los atributos que no corresponden a Lance.
El uso de redes neuronales artificiales, como modelos matemáticos de las redes neuronales biológicas y por ende como modelos del propio cerebro, puede hacer pensar que nuestro programa CODES pretenda ser un modelo mental, lo cual esta muy lejos aún de nuestras intenciones.
Puesto que las redes neuronales son artificios matemáticos que operan sobre números, se hace necesario convertir o codificar las estructuras gráficas a valores numéricos. Solo así podremos relacionar las estructuras con sus propiedades, que normalmente se expresan con valores numéricos.
5. Isomorfismo y - Estructura como Proceso - dentro de la Teoría de la Gestalt
Para solventar este problema hemos recurrido a la Teoría de la Gestalt. Una teoría psicológica, que tuvo su auge a principios del siglo XX. Uno de sus paradigmas más discutidos fue el principio del isomorfismo entre la representación mental y el substrato biológico sobre el que se asienta dicha representación.
En nuestro caso, CODES consta de dos niveles. El primero lo forma un espacio topológico, tal como Turro define formalmente la representación de una estructura. El segundo nivel está constituido por un espacio neuronal, que no es más que la definición formal de una RNA. Si además definimos matemáticamente ambos niveles, tal que los elementos del espacio toplógico se correspondan - uno a uno - con los elementos del espacio neuronal, entonces ambos niveles serán isomorfos. De esta manera hemos creado con CODES, un modelo real del isomorfismo hipotetizado por los teóricos de la Gestalt.
La definición de estructura también ha sido tomada de la teoría de la Gestalt. Köhler entiende como estructura algo más que un simple grafismo. Para él, una estructura es un proceso en el que se interrelacionan todos sus elementos. Este proceso tiende a aproximarse a un estado de equilibrio entre sus elementos, alcanzado éste, se considera que el proceso ha concluido.
El uso de redes neuronales artificiales, como modelos matemáticos de las redes neuronales biológicas y por ende como modelos del propio cerebro, puede hacer pensar que nuestro programa CODES pretenda ser un modelo mental, lo cual esta muy lejos aún de nuestras intenciones.
Puesto que las redes neuronales son artificios matemáticos que operan sobre números, se hace necesario convertir o codificar las estructuras gráficas a valores numéricos. Solo así podremos relacionar las estructuras con sus propiedades, que normalmente se expresan con valores numéricos.
5. Isomorfismo y - Estructura como Proceso - dentro de la Teoría de la Gestalt
Para solventar este problema hemos recurrido a la Teoría de la Gestalt. Una teoría psicológica, que tuvo su auge a principios del siglo XX. Uno de sus paradigmas más discutidos fue el principio del isomorfismo entre la representación mental y el substrato biológico sobre el que se asienta dicha representación.
En nuestro caso, CODES consta de dos niveles. El primero lo forma un espacio topológico, tal como Turro define formalmente la representación de una estructura. El segundo nivel está constituido por un espacio neuronal, que no es más que la definición formal de una RNA. Si además definimos matemáticamente ambos niveles, tal que los elementos del espacio toplógico se correspondan - uno a uno - con los elementos del espacio neuronal, entonces ambos niveles serán isomorfos. De esta manera hemos creado con CODES, un modelo real del isomorfismo hipotetizado por los teóricos de la Gestalt.
La definición de estructura también ha sido tomada de la teoría de la Gestalt. Köhler entiende como estructura algo más que un simple grafismo. Para él, una estructura es un proceso en el que se interrelacionan todos sus elementos. Este proceso tiende a aproximarse a un estado de equilibrio entre sus elementos, alcanzado éste, se considera que el proceso ha concluido.
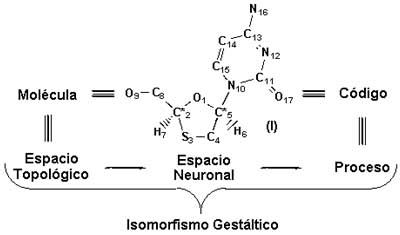
Fig. 5.1. Esquema del isomorfismo: representación y proceso. CODES se fundamenta en la hipótesis de que el pensamiento topológico está fundamentado en representaciones de la geometría topológica. La traslación del espacio topológico abstracto, correspondiente a una molécula, a un espacio neuronal y el subsiguiente procesamiento de la red neuronal así creada, pretende ser una implementación del Isomorfismo Gestáltico. De acuerdo con esta hipótesis, el código generado por CODES, equivale a la representación geométrica (I) que visualiza el espacio topológico correspondiente a la molécula codificada.
Aplicamos ahora esta definición de estructura - como proceso - al espacio neuronal isomorfo a la estructura, cuyos elementos o neuronas van a interaccionar entre sí hasta alcanzar un estado de equilibrio. Alcanzado éste, la red neuronal entra en una fase de reposo y como consecuencia de ello, los valores de la actividad de sus neuronas - una por cada átomo de la molécula - ya no varían. El significado de todo este proceso es que cada uno de estos valores de actividad, es el resultado del equilibrado de las interacciones o influencias con el resto de las neuronas. Este hecho nos permite asignar como equivalente numérico o código de la estructura, a la colección de valores de actividad del conjunto de neuronas de la red neuronal isomorfa. Efectivamente cada uno de estos valores describe, como resultado de la interacción, no sólo el entorno más próximo del correspondiente átomo, sino que también - consecuencia de la interacción múltiple - el entorno más alejado. Lo que en otras palabras hemos definido previamente como representación distribuida. Según esta interpretación toda la información relativa a una estructura esta distribuida uniformente entre todos sus elementos y no localmente. En este último caso la información de cada elemento sería estadísticamente independiente de la de los restantes. Sin embargo, un análisis estadístico del código de la estructura acusa una alta correlación o interdependencia entre sus elementos.
Como queda dicho, para cada estructura, CODES genera un vector numérico o vector estático con tantos elementos como átomos tiene la molécula. Puesto que el procesamiento de esta red neuronal artificial es secuencial o en otras palabras, discontinuo, dispondremos para cada subproceso o iteración, un vector de actividades. Este conjunto de vectores, de los cuales el último es el ya citado vector estático, describe por entero el proceso que ha seguido la red desde su estado inicial, hasta alcanzar el estado final o de equilibrio. Si agrupamos este conjunto de vectores en una matriz obtendremos la que denominamos matriz dinámica puesto que en ella se recogen las modificaciones sufridas por la RNA isomorfa, durante la totalidad del proceso. En definitiva la codificación de una estructura mediante CODES, proporciona dos tipos de datos, el primero sería el vector estático que no es más que una instantánea del estado final o de equilibrio del proceso, mientras que la matriz dinámica nos refleja una secuencia de la totalidad del proceso.
6. Validación de CODES
Una vez que hemos codificado las estructuras - surge la duda razonable - si los valores numéricos generados por CODES realmente representan a la estructura de partida. Para ello transponemos la matriz dinámica de una molécula, e identificamos cada una de sus filas con el nombre de la unidad o átomo presente en la red neuronal isomorfa. Esta matriz transformada, se procesa mediante una red neuronal de Kohonen, que reduce la compleja dimensionalidad de la misma a sólo dos dimensiones, que ya pueden visualizarse gráficamente en un sistema de coordenadas. Lo que muestran estos mapas de Kohonen, no es más que la distribución en un espacio bidimensional de las unidades o átomos de la red neuronal isomorfa, conjuntamente con su entorno interactivo, que queda de manifiesto cuando se analizan los pesos que ligan a las unidades o átomos entre sí. Así valores positivos altos representan valores de interacción positiva elevada, mientras que pesos negativos señalan interacciones negativas. Este entorno interactivo reproduce exactamente el entorno de enlace (interacción positiva) y no enlace químico (interacción negativa) que tiene cada átomo de la molécula con respecto a sus átomos vecinos.
Aplicamos ahora esta definición de estructura - como proceso - al espacio neuronal isomorfo a la estructura, cuyos elementos o neuronas van a interaccionar entre sí hasta alcanzar un estado de equilibrio. Alcanzado éste, la red neuronal entra en una fase de reposo y como consecuencia de ello, los valores de la actividad de sus neuronas - una por cada átomo de la molécula - ya no varían. El significado de todo este proceso es que cada uno de estos valores de actividad, es el resultado del equilibrado de las interacciones o influencias con el resto de las neuronas. Este hecho nos permite asignar como equivalente numérico o código de la estructura, a la colección de valores de actividad del conjunto de neuronas de la red neuronal isomorfa. Efectivamente cada uno de estos valores describe, como resultado de la interacción, no sólo el entorno más próximo del correspondiente átomo, sino que también - consecuencia de la interacción múltiple - el entorno más alejado. Lo que en otras palabras hemos definido previamente como representación distribuida. Según esta interpretación toda la información relativa a una estructura esta distribuida uniformente entre todos sus elementos y no localmente. En este último caso la información de cada elemento sería estadísticamente independiente de la de los restantes. Sin embargo, un análisis estadístico del código de la estructura acusa una alta correlación o interdependencia entre sus elementos.
Como queda dicho, para cada estructura, CODES genera un vector numérico o vector estático con tantos elementos como átomos tiene la molécula. Puesto que el procesamiento de esta red neuronal artificial es secuencial o en otras palabras, discontinuo, dispondremos para cada subproceso o iteración, un vector de actividades. Este conjunto de vectores, de los cuales el último es el ya citado vector estático, describe por entero el proceso que ha seguido la red desde su estado inicial, hasta alcanzar el estado final o de equilibrio. Si agrupamos este conjunto de vectores en una matriz obtendremos la que denominamos matriz dinámica puesto que en ella se recogen las modificaciones sufridas por la RNA isomorfa, durante la totalidad del proceso. En definitiva la codificación de una estructura mediante CODES, proporciona dos tipos de datos, el primero sería el vector estático que no es más que una instantánea del estado final o de equilibrio del proceso, mientras que la matriz dinámica nos refleja una secuencia de la totalidad del proceso.
6. Validación de CODES
Una vez que hemos codificado las estructuras - surge la duda razonable - si los valores numéricos generados por CODES realmente representan a la estructura de partida. Para ello transponemos la matriz dinámica de una molécula, e identificamos cada una de sus filas con el nombre de la unidad o átomo presente en la red neuronal isomorfa. Esta matriz transformada, se procesa mediante una red neuronal de Kohonen, que reduce la compleja dimensionalidad de la misma a sólo dos dimensiones, que ya pueden visualizarse gráficamente en un sistema de coordenadas. Lo que muestran estos mapas de Kohonen, no es más que la distribución en un espacio bidimensional de las unidades o átomos de la red neuronal isomorfa, conjuntamente con su entorno interactivo, que queda de manifiesto cuando se analizan los pesos que ligan a las unidades o átomos entre sí. Así valores positivos altos representan valores de interacción positiva elevada, mientras que pesos negativos señalan interacciones negativas. Este entorno interactivo reproduce exactamente el entorno de enlace (interacción positiva) y no enlace químico (interacción negativa) que tiene cada átomo de la molécula con respecto a sus átomos vecinos.
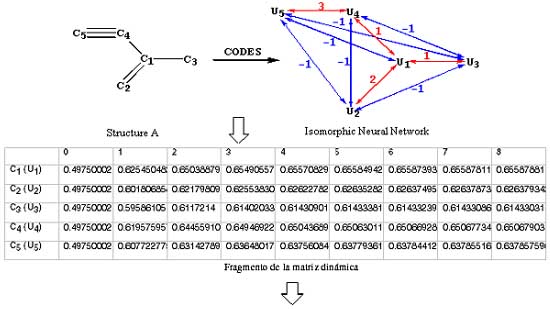
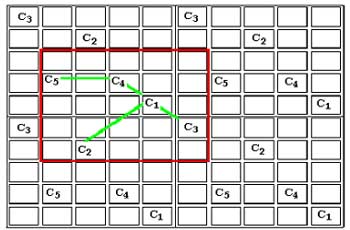
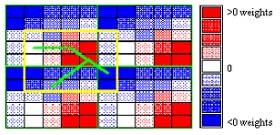
Fig. 6.1. Esquema en el que se representa el proceso de codificación de la estructura A y su validación. Se visualiza la red neuronal isomorfa con los distintos tipos de conexiones entre sus unidades o neuronas. Su procesamiento de lugar al código, en éste caso representado por un fragmento de la matriz dinámica. Esta matriz recoge los valores de la actividad de las unidades o neuronas de la red neuronal isomorfa durante cada una de las iteraciones hasta llegar al estado de equilibrio. La validación del código resultante se evidencia con la ayuda de un mapa de Kohonen alimentado con la matriz dinámica de CODES. El mapa de Kohonen y su distribución de pesos, nos muestra que éstos se corresponden con la resultante de los pesos de las conexiones de la red que inciden sobre cada unidad o neurona. Estas conexiones a su vez, se corresponden con los enlaces de la molécula.
7. Interpretación de la isomería óptica mediante CODES
7. Interpretación de la isomería óptica mediante CODES
Fig. 7.1. Las señales X1, X2, X3 y X4, que llegan a una unidad o neurona correspondiente a un centro quiral, son transformados no linealmente. Estas actividades provienen de los cuatro sustituyentes del centro quiral y se presentan ordenadas en función de su magnitud. Esta ordenación ha permitido asignar previamente una configuración R o S al centro quiral. En función de esa asignación se transforman no linealmente las señales de entrada X1, X2, X3 y X4,, de acuerdo con la función representada en la Fig. 7.3.
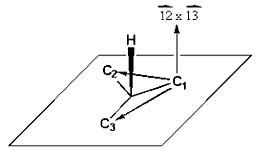
Fig. 7.2. El signo del producto vectorial en combinación con la orientación del enlace estereoquímico significativo, conducen a la asignación interna del centro quiral (R o S).
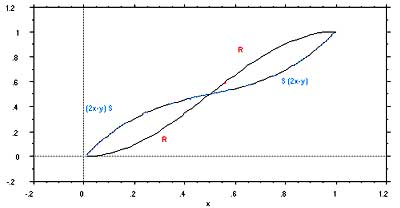
Fig. 7.3. Representación de las funciones mediante las que se transforman - no linealmente - las entradas a un centro quiral.
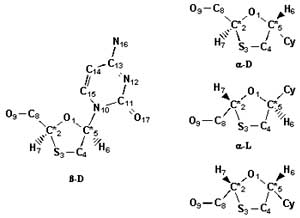
Fig. 7.4. Estructuras de los cuatro isómeros correspondientes a la sustitución de citosina en el C5 del anillo de 1,3-oxatiolano. La muestra completa esta integrada por 76 estructuras, agrupadas en 19 conjuntos correspondientes cada uno a una base púrica o pirimidínica y que constan de 4 isómeros equivalentes a los de la figura. Este estudio ha servido para comprobar como se pueden clasificar estas 76 moléculas de acuerdo con su estructura tridimensional, partiendo únicamente de dos descriptores (H6 y H7) obtenidos por CODES. Hay que resaltar que la información que procesa CODES es estrictamente topológica y por tanto adimensional.
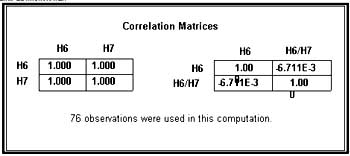
Fig. 7.5. Valores de correlacion de H6 y H7.
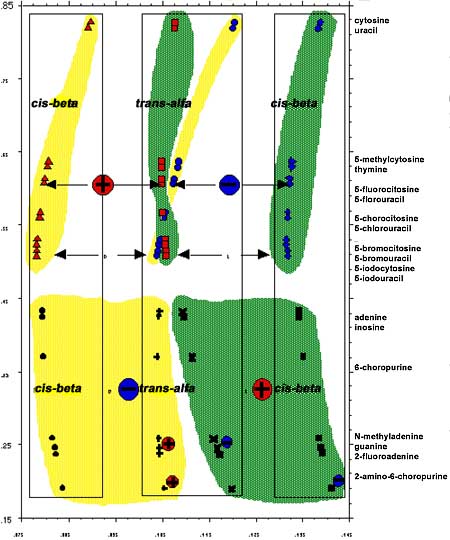


Fig. 7.6. Clasificación de la muestra de 76 pseudo nucleósidos de acuerdo con la naturaleza de la base (eje H6) y de los dos centros quirales (eje H6/H7), presentes en el anillo de 1,3-oxatiolano. Las zonas coloreadas agrupan las estructuras con isomería trans en los alfa-D y alfa-L y cis en los beta-D y beta-L.
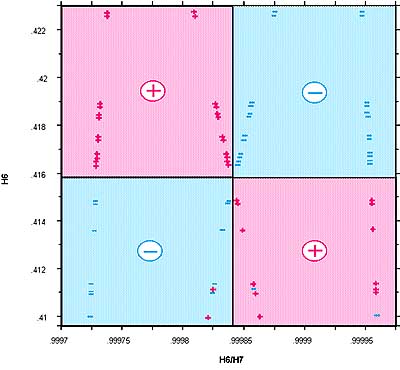
Fig. 7.7. Distribución por cuadrantes de los signos del poder rotatorio óptico de 76 pseudo nucleósidos. Atención a las dos parejas de compuestos que no cumplen esta regla. Mediante una modificación de la función representada en la Fig. 7.3 se ha logrado mejorar la resolución de las isómeros representados en la parte central de la Fig. 7.6.
8. Predicción y Validación mediante RNA de Retropropagación del Error
Confirmada así la validez de los códigos numéricos generados por CODES, sólo resta ya por establecer las relaciones entre estructuras conocidas y sus propiedades, como primer paso antes de pasar a predecir las propiedades de nuevas estructuras. Todas estas operaciones se han efectuado con Redes Neuronales Artificiales del tipo de Retropropagación del Error. Su funcionamiento no consiste más que en la asociación de los pares de datos - código y actividad biológica - que corresponden a cada molécula del conjunto de la muestra. Al cabo de un suficiente número de iteraciones, la RNA acaba por aprender qué actividad biológica le corresponde a cada código o estructura. Este proceso equivale a la creación de una base de datos o memoria ya mencionada con anterioridad. La RNA así entrenada, es capaz de asignar o predecir la actividad biológica de una nueva estructura, distinta de las que han servido para su entrenamiento y por tanto desconocida para la RNA. El proceso de generalización - implícito en la RNA - es capaz de extraer esta información del conocimiento distribuido por todas las unidades o neuronas de la RNA, que ésta previamente ha adquirido durante el entrenamiento.
La validez del resultado de estas predicciones se comprueba mediante el método conocido como "deja uno fuera" (leave one out ó LOO) que consiste en repetir el entrenamiento, tantas veces como moléculas tiene la muestra, dejando en cada caso una de las moléculas fuera. La actividad biológica de esta molécula y de las restantes, es posteriormente predicha mediante cada una de las RNA entrenadas. El ajuste de estos valores de actividad predichos con los valores conocidos, nos dará una idea de la calidad del proceso de predicción.
Como ya ha quedado dicho, éstas redes una vez entrenadas con una colección de estructuras y su correspondiente propiedad, se convierten en una suerte de memoria o base de conocimiento, capaz de generalizar o extender su conocimiento a estructuras nuevas, condición indispensable para dar lugar a las correspondientes predicciones de las propiedades escogidas (mecanismo de acción de un fármaco, punto de fusión, olor o sabor de una molécula, etc).
Otra de las aplicaciones de las RNA de Retropropagación del Error - en forma de Red Autoasociativa - ha sido la reducción de dimensiones de los códigos de CODES (vector estático o matriz dinámica), puesto que cada uno de ellos tiene tantas dimensiones como átomos tiene la molécula de origen. Esta operación se hace necesaria para homogenizar los datos de un conjunto de moléculas, puesto que la dimensionalidad del vector de entrada o input a una red de Retropropagación ha de ser la misma para todo el conjunto de la muestra.
En la actualidad estamos utilizando la matriz dinámica como entrada a una Red de Retropropagación del Error del tipo TimeDelay (TDNN). Estas redes son capaces de aprender procesos dependientes del tiempo. Los resultados obtenidos son especialmente llamativos.
Agradecimientos
No quiero terminar esta exposición sin agradecer a la AEC, la distinción con la que me ha honrado. Esta distinción, resulta especialmente de agradecer por el carácter acusadamente interdisciplinar que tiene la labor aquí expuesta. Tampoco quiero dejar de citar aquí, a todos aquellos, que a lo largo de mucho tiempo, han contribuido al desarrollo de CODES y con su colaboración y entusiasmo han demostrado creer en este proyecto:
8. Predicción y Validación mediante RNA de Retropropagación del Error
Confirmada así la validez de los códigos numéricos generados por CODES, sólo resta ya por establecer las relaciones entre estructuras conocidas y sus propiedades, como primer paso antes de pasar a predecir las propiedades de nuevas estructuras. Todas estas operaciones se han efectuado con Redes Neuronales Artificiales del tipo de Retropropagación del Error. Su funcionamiento no consiste más que en la asociación de los pares de datos - código y actividad biológica - que corresponden a cada molécula del conjunto de la muestra. Al cabo de un suficiente número de iteraciones, la RNA acaba por aprender qué actividad biológica le corresponde a cada código o estructura. Este proceso equivale a la creación de una base de datos o memoria ya mencionada con anterioridad. La RNA así entrenada, es capaz de asignar o predecir la actividad biológica de una nueva estructura, distinta de las que han servido para su entrenamiento y por tanto desconocida para la RNA. El proceso de generalización - implícito en la RNA - es capaz de extraer esta información del conocimiento distribuido por todas las unidades o neuronas de la RNA, que ésta previamente ha adquirido durante el entrenamiento.
La validez del resultado de estas predicciones se comprueba mediante el método conocido como "deja uno fuera" (leave one out ó LOO) que consiste en repetir el entrenamiento, tantas veces como moléculas tiene la muestra, dejando en cada caso una de las moléculas fuera. La actividad biológica de esta molécula y de las restantes, es posteriormente predicha mediante cada una de las RNA entrenadas. El ajuste de estos valores de actividad predichos con los valores conocidos, nos dará una idea de la calidad del proceso de predicción.
Como ya ha quedado dicho, éstas redes una vez entrenadas con una colección de estructuras y su correspondiente propiedad, se convierten en una suerte de memoria o base de conocimiento, capaz de generalizar o extender su conocimiento a estructuras nuevas, condición indispensable para dar lugar a las correspondientes predicciones de las propiedades escogidas (mecanismo de acción de un fármaco, punto de fusión, olor o sabor de una molécula, etc).
Otra de las aplicaciones de las RNA de Retropropagación del Error - en forma de Red Autoasociativa - ha sido la reducción de dimensiones de los códigos de CODES (vector estático o matriz dinámica), puesto que cada uno de ellos tiene tantas dimensiones como átomos tiene la molécula de origen. Esta operación se hace necesaria para homogenizar los datos de un conjunto de moléculas, puesto que la dimensionalidad del vector de entrada o input a una red de Retropropagación ha de ser la misma para todo el conjunto de la muestra.
En la actualidad estamos utilizando la matriz dinámica como entrada a una Red de Retropropagación del Error del tipo TimeDelay (TDNN). Estas redes son capaces de aprender procesos dependientes del tiempo. Los resultados obtenidos son especialmente llamativos.
Agradecimientos
No quiero terminar esta exposición sin agradecer a la AEC, la distinción con la que me ha honrado. Esta distinción, resulta especialmente de agradecer por el carácter acusadamente interdisciplinar que tiene la labor aquí expuesta. Tampoco quiero dejar de citar aquí, a todos aquellos, que a lo largo de mucho tiempo, han contribuido al desarrollo de CODES y con su colaboración y entusiasmo han demostrado creer en este proyecto:
- Dra. Carmela Ochoa de Ocáriz
Dra. Ana Martínez Gil
Dr. Juan Rodriguez Ordóñez
Mª Mercedes Rodríguez Fernández
Antonio Chana López
Joaquín Cáceres Reyes